Hacia una Nueva Fisiopatología de las Osteopatías Fragilizantes
José L Ferretti11 Centro de Estudios del Metabolismo Fosfocalcio (CEMFoC), Universidad Nacional de Rosario, Rosario, Argentina
Resumen
Palabras Clave: remodelación ósea, mecanostato, osteopenia, densitometría
Dentro de los limites fisiológicos, la fragilidad del tejido óseo esta condicionada al grado de sufrimiento mecánico que le provocan las cargas a que se ve sometido por el uso o por los traumas. Ese sufrimiento esta directa y exclusivamente determinado por la cantidad de deformación provocada en cada caso. En consecuencia, con la sola excepción de estados extremos como la osteoporosis, la integridad física de los huesos esta determinada fundamentalmente por la rigidez que les confiere su estructura en cada región, en relación con el sentido de deformación que se considere (9, 29).
No debe extrañar, entonces, que el tejido óseo sea una estructura nativamente biomecánica (5, 18, 21, 29, 45), orientada al logro de una “rigidez típica”.Esta esta definida por un límite máximo de 2 milésimas de deformación, en cualquier dirección, ocasionada por las cargas máximas habituales, para todas las regiones de todos los huesos de todos los vertebrados (45). Esta propiedad permitió que la materia viva pudiera evolutivamente “salir del agua”, autoportándose y manteniendo su homeostasis mineral; y todavía se mantiene estrictamente controlada en todas las especies y para todos los huesos, con excepción de la calota craneana, el hueso turbinal y los osículos del oído (35). Se esta empezando a dilucidar el mecanismo de control homeostático de la eficiencia (o “función de sostén”) esquelética (1, 10, 18, 20, 21, 29, 34-39, 41). Las células intrínsecas de la estructura ósea resistiva (osteocitos) están especializadas para detectar las deformaciones producidas (Figura 1, izquierda), a las cuales respondería orientando direccionalmente su liberación de mediadores paracrinos, que estimulan o inhiben la formación o la destrucción de huesos por partes de las células extrínsecas al tejido duro (osteoblastos y osteoblastos) (1, 41, 45).
Así, un aumento de la tasa de deformación máxima habitual (ganancia de peso, actitud física, ejercicio) induce a las “lining cells” regionales (osteoblastos inactivos que recubren el material calcificado) a activarse y agregar material óseo (Figura 1, centro). Opuestamente, una reducción de la tasa de deformación máxima por el uso (perdida de peso, inactividad, ingravidez) induce una de las de las “lining cells” locales, que descubren el material calcificado y determina su destrucción osteoclástica (Figura 1, derecha) Mediadores liberados del tejido destruido podrán o no inducir un ulterior acoplamiento formativo osteoblástico, construyéndose una Unidad Multicelular Ósea (BMU).
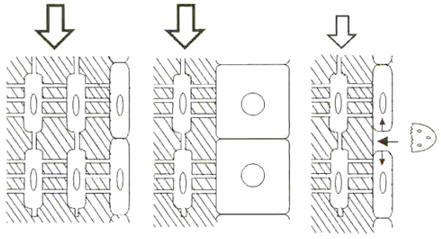
Figura 1. Esquema elemental de interacciones de carga (Fechas: normal a
la izquierda; aumentada al centro; reducida a la derecha) (1, 39).
El conjunto de procesos de formación y destrucción ósea no acopladas se denominan modelación; es responsable de la adquisición de la forma y el tamaño óseos; con balance global usualmente positivo, es el único recurso conocido que permite ganar masa ósea, y lamentablemente declina con la edad. Cuando tiene lugar el acoplamiento localizado destrucción/formación (BMU), el proceso se denomina remodelación. La remodelación coadyuva a un 30% de la homeostasis mineral; es el único recurso conocido para recambiar el material óseo y para reparar microfracturas, y no declina con la edad. La remodelación puede operar según dos modalidades: respondiendo el material removido en forma total (como “conservativo”), o parcial (modo “desuso”). La remodelación en modo “desuso” es el único mecanismo posible para perder masa ósea, y la única de todas las osteopenias y las osteoporosis conocidas (4, 6, 34-39, 48).
La modelación y remodelación, actuando en cada región estética puede producir balances positivos (fisiológicos en la juventud) o negativos (fisiológicos luego de la adultez) de masa mineral que pueden afectar aunque en forma no obligadamente proporcional la eficiencia mecánica ósea. La masa mineral y la eficiencia mecánica ósea son independientes, por que esta última no esta determinada solo por la cantidad, sino también por la calidad (rigidez intrínseca) y la distribución especial del material duro (18, 20, 21, 29,42) (Tabla 1).

Tabla 1. Determinantes de la calidad de un hueso integrado y de sus dos
determinantes: la calidad mecánica del material óseo y la calidad del diseño
macroarquitectónico de la estructura.
Estas características esqueléticas están determinadas por un sistema regulatorio: el “mecanostato óseo” (18, 20, 21, 34, 39) (Figura 2). El macanostato tiende a mantener constante la referida “rigidez típica” (2 milésimas de deformación) para las direcciones habituales de las cargas máximas impuestas por el uso (generalmente derivado de las contracciones musculares) en todas las regiones esqueléticas. De esta manera, queda determinado que “los huesos tiende a ser estructuralmente lo que la musculatura regional quiere” (35, 46). La Figura 3 muestra una relación lineal única entre la cantidad ósea (topográfica) y la fuerza musculares regional (dinamométrica) humanas, independiente del sexo y la edad.
Además de estas interrelaciones homeostáticas, la actividad de todas las células óseas esta permanentemente modulada (no “regulada” o “controlada”) por el ambiente endo/paraclinico-metabólico. A diferencia del macanostato, estos factores sistémicos estimulados inhiben no direccionalmente la moderación y la remodelación en todas partes, con total indiferencia de las necesidades mecánicas regionales esqueléticas.
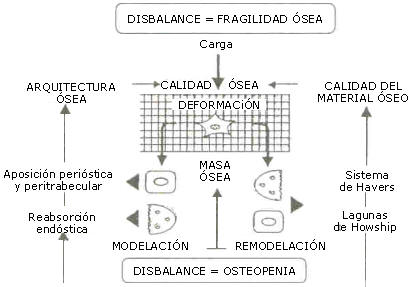
Figura 2. Esquema elemental del mecanostato óseo, centrado en el control
ostecítico de la deformabilidad tisular local, que distingue disbalances de masa
(abajo) y de calidad mecánica (arriba), como entidades etipatogénicamente
independientemente.
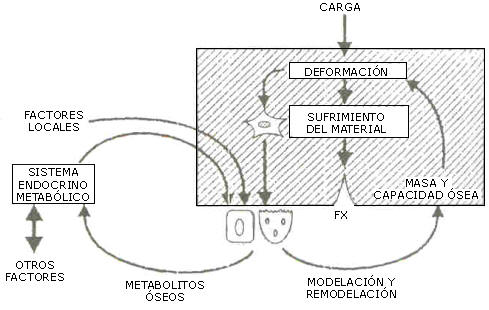
Figura 3. Perturbación endócrino-metabólica del trabajo regulatorio
mecanostático de la calidad estructural esquelética. No hay tasa de
retroalimentación que informe variaciones de masa o de calidad ósea (31-33).
El normal funcionamiento del mecanostato requiere ineludiblemente la normalidad de todo el aparato endo/paracrino-metabólico. En esas condiciones, el mecanostato requiere paraclinico-metabólico. Es esas condiciones, el macanostato bastaría por si solo para controlar la deformabilidad (y con ella la eficiencia mecánica) de la estructural ósea. Sin embargo, el rendimiento del trabajo del mecanostato esta supeditado a los requerimientos de los sistemas regulatorios de la homeospatasis mineral del medio interno. Como todos los sistemas de control humorales, ellos son filogenéticamente anteriores a la “salida del agua” de las especies, y vitales para cualquier organismo pluricelular, incluso para la función de las mismas células óseas. Esta circunstancia ha subordinado la eficiencia del mecanostato a los avatares del control de las “señales de error” de esos sistemas (concentraciones sanguíneas de metabolitos y hormonas-madre), que nada tiene que ver con la regulación de la masa (que no existe) ni de la eficiencia mecánica ósea. (Figura 4).
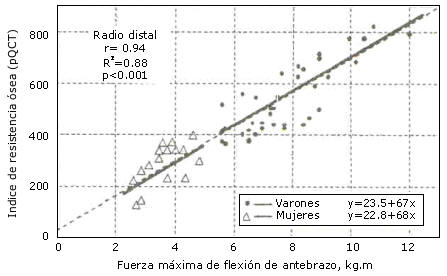
Figura 4. Demostración de que más del 80% (r2) de la calidad ósea
del radio distal humano [estimado por un índice tomográfico de resistencia (25,
45)] esta linealmente determinada por la fuerza dinamométrica de flexión del
antebrazo, con independencia del sexo y la edad (45).
Curiosamente, en vez de construir a manera estable la “deformación típica” ósea (punto de referencia o “set-point” del mecanostato), los sistemas de control de la homeostasis mineral tienden a modificarla permanentemente sin respetar direccionalidad ni regionalidad; es decir, “desorientando” el trabajo de control biomecánico de la eficiencia estructural esquelética (2, 7, 8, 12 , 15 , 17 , 22 , 23 , 35 , 40). Por esta razón, los sistemas endocrino-metabólicos aun cuando su equilibrio resulte primordial para el normal funcionamiento de las células óseas, se comportan fisiológica o patológicamente como perturbadores (nunca reguladores) de la integridad funcional esquelética (29, 35, 36, 38). Contrariamente a lo que pudiera suponer por intuición, ninguna hormona regula (en el estricto sentido cibernético del término) ninguna propiedad esquelética. No existe ningún factor humoral conocido que facilite, dificulte, provoque o impida, con sentimiento regulatorio local, las ganancias de masa y calidad estructural derivadas del ejercicio; ni las pérdidas producidas por inactividad, inmovilización o ingravidez (que pueden llegar al 60% de la masa mineral en pocos meses). Tampoco se conoce vector alguno de retroalimentación que, con punto de partida en el esqueleto, informe a las glandulas endócrinas de los resultados estructurales de su acción perturbadora, ni del estado actual de la masa o la eficiencia mecánica ósea (Figura 3). Obviamente. No se puede regular lo que no se conoce.
Cualquier anormalidad de la fragilidad ósea implicará naturalmente alguna clase de desperfecto en el trabajo de control mecanostático (de otra forma, la misma no podría tener lugar). En consecuencia, los mecanismos etiopatogénicos de todas las osteopatías fragilizantes (incluyendo la “osteoporosis”) se pueden restringir a las dos únicas maneras posibles de alterar el trabajo de control del mecanostato: (1) reproducir su “input” (descarga, por déficit local de uso mecánico); o (2) desplazar su “set-point” (desorientación, por perturbación sistémica no compensada; Figura 5) (36-38).
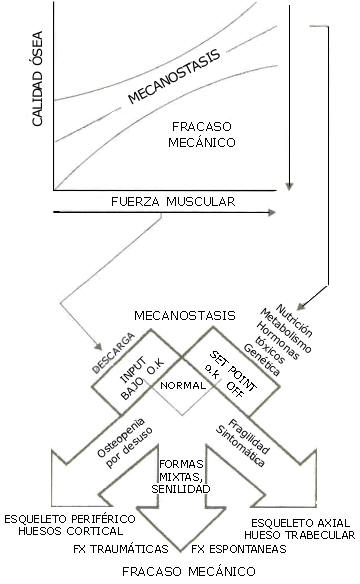
Figura 5. Concepción mecanostática de la etiopatiogenia de las
osteopatías fragilizantes (34). La parte superior a la figura esta asociada con
la correlación en la Figura 4.
En el primer caso se producirá y una osteopenia (no “osteoporosis”) por desuso que afectará predominantemente al hueso cortical y al esqueleto periférico, tornado a los huesos relativamente frágiles respecto de los esfuerzos normales para un sujeto sano, pero adecuados al trabajo habitual del portador. El tratamiento de esta condición fisiológica deberá limitarse a recargar mecánicamente en sistema. En el segundo caso, tendrá lugar una fragilidad ósea “sintomática” (de la condición que originó el desequilibrio), que afectará principalmente al esqueleto axial, reduciendo la resistencia de los huesos a los esfuerzos habituales del portador. El tratamiento de esta enfermedad es la eliminación del factor que desplazó el “set-point” del mecanostato (dis-endocrina, desnutrición, decaimiento de receptores, etc.). La importancia patogénica de los sistemas de control endocrinos-metabólicos a este respecto es evidente, ya que su desequilibrio constituye la causa de la enorme mayoría de los síndromes osteoporóticos conocidos. Combinaciones de ambas etiologías pueden producir casos intermedios con características mixtas, como la fragilidad ósea senil (37).
Esta concepción plantea la necesidad de completar el diagnóstico corriente de “osteoporosis”, adjudicándole el correlato biomecánico del cual hoy carece (4,48). El término “osteoporosis”, definido sólo “cuantitivamente” por un t-score densitométrico inferior al “límite osteopénico” (¿?) de -2.5 y “ establecido” (¿?) por la ocurrencia de fracturas (11, 39), quedaría automáticamente excluido de este esquema. No podría aceptarse, de acuerdo con lo expuesto, que la “osteoporosis” sea simplemente una “osteopenia intensa”; o que ningún “osteopénico fracturado” (con t-score entre -1.0 y -2.5) sea osteoporótico, como surgiría de la aplicación estricta de esas pautas de diagnóstico (Figura 6).
La aceptación del diagnóstico cuantitativo de “osteoporosis” (explicable por el momento, a falta de mejor recurso [39]) viene fundamentada por la reiterada demostración de correlaciones significativas entre masa mineral densitométrica (BMC, BMD) y resistencia mecánica o riesgo de fractura. Esas correlaciones, pese a resultar estadísticamente válidas en términos poblacionales, son espúreas (43), Marshall, Francés; es decir, no permite inferir asociaciones causales directas entre masa mineral y calidad mecánica para cada individuo. Esto es así por que la masa ósea está subrogada a los verdaderos determinantes de la resistencia mecánica ósea, que son la calidad mecánica y la distribución espacial del material calcificado. Negarlo implica aceptar que el simple aumento de una masa ósea reducida corregirá la fragilidad en cada persona, y está demostrado en estudios empleando flúor, calcitonina y bisfosfonatos, que eso no ocurre obligadamente así.
Subsanar esta deficiencia diagnostica tendría implicancias terapéuticas muy importantes, porque nos permitiría calificar adecuadamente los verdaderos efectos de los tratamientos corrientes sobre las propiedades óseas relevantes (reducir la fragilidad esquelética en las regiones deficitarias) (4, 6, 16, 19, 24, 27, 40, 44, 48). Poseemos, a esos efectos, medicamentos estimulantes moderatorios e inhibidores remodelatorios (no “inhibidores de la reabsorción” en forma pura, como se los llama). Esto puede provocar la ilusión de contar con un arsenal terapéutico suficiente. Sin embargo, los conocimientos actuales sobre las interacciones regulatorias expuestas, nos obligan a repensar algunos aspectos fundamentales del problema (34-38):
- De a poco nada sirve engrosar estructuras trabeculares desconectadas, o ubicadas en sitios mecánicamente irrelevantes para la determinación de la iniciación del trazo fracturario (como el interior del cuello femoral o del radio).
- Agregar material del lado intracortical diafisario (que contribuye poco a mejorar el momento de inercia de la sección ósea); así como colocar, en el lugar adecuado, un material nuevo de baja calidad mecánica.
- Tampoco ayudaría inhibir la sustitución o la eliminación remodelatoria fisiológica del material óseo mecánicamente inadecuado, dificultando a cambio, la homeostasis mineral y la reparación de microfacturas del material mecánicamente efectivo en otras regiones.
- Además, de nada valdría el logro de cualquier mejoría biomecánica efectivamente demostrable, si al suspender el tratamiento el mecanostato volviera a la situación anterior de descontrol, porque entonces el mismo se encargaría de neutralizar en poco tiempo todo el balance positivo obtenido, en todos los casos (35,36).
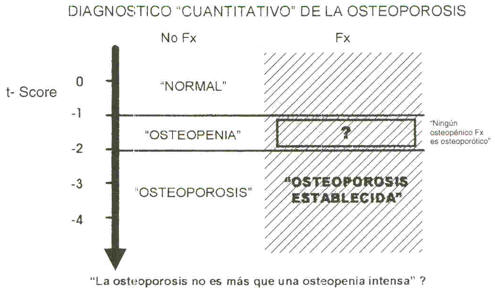
Figura 6. Incongruencias del criterio diagnostico “cuantitativo”
actualmente aceptado para osteopenias y osteoporosis, discutido en el texto
(36).
Si no se pueden suprimir definitivamente las causas de la salida de cauce del mecanostato, el único camino para el éxito del tratamiento sería “reencauzar” permanentemente al sistema hacia la producción de hueso mecánicamente efectivo, en la región considerada. Todavía no contamos con recursos que nos permitan esgrimir esa pretensión.
Una buena manera de comenzar a evitar estos peligros es reconocer que el esqueleto no es una masa endócrinamente regulada por efectos hormonales sobre blastos y clastos, con “un cierto correlato” biomecánico, que nace, crece, llega a un máximo y luego decae hasta reducir críticamente su resistencia, como intuitivamente se tiende a aceptar (12, 39) (Figura 7., izquierda). Por el contrario, cada región ósea es una estructura constituida por un material anisotrópico (de comportamiento supeditado a la dirección de las deformaciones inducidas por las cargas habituales), espacialmente orientado para lograr una cierta rigidez típica por un sistema regulatorio esencialmente mecánico (el mecanostato) a punto de partida osteocítico, potencialmente autosuficiente, pero constantemente perturbado por acciones sistémicas sobre sus efectores (blastos y clastos; Figura 7, derecha) (4-6, 18, 20, 21, 29, 34-38, 48).
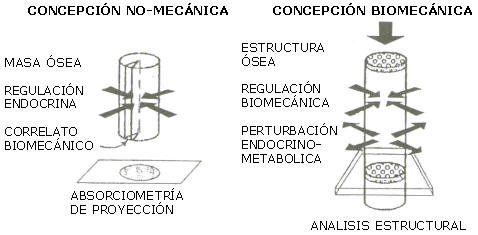
Figura 7. Concepciones no biomecánica (“Paradigma de los años ‘60”) y
biomecánica (“Paradigma de Utah”) de la naturaleza estructural ósea (33).
Esto ayudará a comprender que, cualquiera sea su masa, cualquier región esquelética puede llegar a perder su resistencia a microfracturarse ante deformaciones críticas, con el solo requisito de que la calidad mecánica y/o la distribución espacial de su tejido calcificado sean inadecuadas a las cargas máximas localmente impuestas por el uso habitual. Del mismo modo, quedará claro que no será la masa, sino la calidad del material y la eficiencia arquitectónica del diseño lo que habrá que corregir para recobrar la normalidad mecánica de la estructura en la región afectada (4, 6, 48).
Así para cuantificar la fragilidad ósea, debemos completar las determinaciones densitometrías de masa (ideales para el diagnostico de osteopenia, pero no de “osteoporosis”) con análisis estructurales no-invasivos de la calidad mecánica y la distribución espacial del material calcificado, en la región que se desea estudiar (no en otra) y de acuerdo con la dirección de las cargas causantes de las deformaciones que inducirían su fractura (no con otra, o con ninguna dirección; Figura 8).
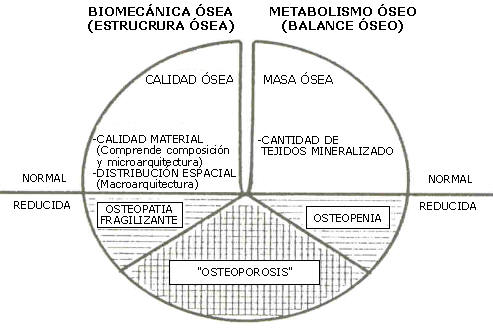
Figura 8. Diagnóstico combinado, metabólico (relativo al balance de la
masa) y biomecánico (relativo al balance de calida estructural), de las
osteopenias simples y las osteopatías fragilizantes (26, 27, 34).
Buena parte de ésto se estima posible aplicando combinaciones de métodos novedosos, actualmente en desarrollo (26, 28, 31) (Tabla 2). La tomografía computada axial (QCT) o periférica (pQCT) puede determinar muy bien ciertos parámetros arquitectónicos y medir la densidad mineral volumétrica del tejido cortical sólido, que estima al menos un componente (el mineral) de la calidad mecánica del material óseo (25, 28, 30, 31, 33, 46, 47). La ultrasonometría puede determinar globalmente la calidad mecánica (rigidez intrínseca) del tejido cortical “sólido” (tibial), que se estima bastante constante en todo el esqueleto para un mismo individuo. La misma densitometría, como alternativa de técnicas tomográficas o dinamométricas más específicas que deberían estandardizarse adecuadamente, podría contribuir a evaluar con esta finalidad el estado de la musculatura regional por su condición de “masa magra” a efectos correlativos (31, 32).
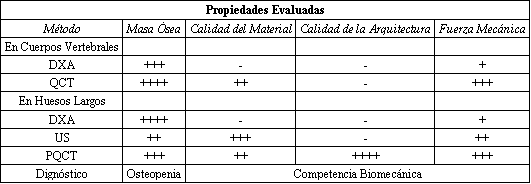
Tabla 2. Metodología disponible (unidades relativas expresadas en cruces)
para intentar el diagnóstico funcional, biomecánico, de las osteopenias y las
osteopatías fragilizantes (29-31). (US = Ultrasonometría).
Le mejor manera de diferenciar osteopenias de osteoporosis es tener en cuenta la relación entre calidad ósea regional real del individuo, y la que necesitaría tener para soportar las cargas máximas habituales de acuerdo con su fuerza muscular regional determinada o estimada (4, 48); esto se denomina “factor de seguridad” (“safety factor”) (3), y debería idealmente valer 1. Distinguiríamos así dos entidades clínicas esencialmente distintas (37):
- “Osteopenia simple”, o “por desuso”: reducción de masa ósea sin compromiso biomecánico, con mecanostasis controlada y (“safety factor”) igual o mejor que 1 para el hueso y la musculatura disponibles, para cuyo diagnóstico la densitometría es y será por mucho tiempo el mejor recurso conocido, especialmente si también se la emplea para determinar masa muscular (31, 32) (Figura 5, arriba, zona de “mecanostasis”, y abajo, izquierda).
- “Fragilidad ósea sintomática”: reducción de la calidad mecánica ósea por debajo de la proporción normal con la fuerza muscular regional (riesgo de fractura aumentado para cargas habituales máximas en la región), que podrá ser tanto “osteopénica” como “no-osteopénica), con (“safety factor”) menor que 1, independientemente del valor absoluto de masa ósea, y para cuyo diagnóstico necesitaríamos las tecnologías complementarias (Figura 5, arriba, zona de “fracaso mecánico”, y abajo, derecha).
La “osteoporosis”, quedaría así comprendida (en forma no excluyente) dentro de los estados de fragilidad aumentada osteopénica, definida y diagnosticada como una “osteopenia con correlato biomecánico” (Figura 8) determinada por un desplazamiento patológico del “set-point” del mecanostato (4,8, 37, 38) (Figuras 2, 4, 5), que conlleva riesgo fracturario aumentado a las tasas de carga habituales para el sujeto, es decir, en relación con la aptitud de su musculatura regional (“safety factor” menor que 1), sin otra alusión fisiopatológica.
Referencias
1. Aarden EM, Burger EH, Nijweide PJ (1994). Function of osteocytes in bone. J Cell Biochem 55: 287-299
2. Biewener AA (1993). Safety factors in bone strength. Calc Tiss Int 53(S): 68-74
3. Burr DB, Martin RB (1989). Errors in bone remodeling: toward a unified theory of mechanical bone disease. Am J Anat 186: 186-216
4. Burr DB, Martin RB (1992). Mechanisms of bone adaptation of the mechanical environment. Triangle (Sandoz) 31:59-76
5. Burr DB (1997). Muscle strength, bone mass, and age-related bone loss. J Bone Miner Res 12:1547-15514
6. Capozza RF, Ferretti JL, Ma YF, Meta M, Alippi R, Zanchetta JR, jee WSS (1995). Tomografic (pQCT) and biomechanical effects of hPTH (1-38) on chonically immobilized overloaded rat femur. Bone 17:S233-S239
7. Cointry GR, Mondelo N, Zanchetta JR, Montuori E, Ferretti JL (1995). Intravenous olpadronate restores ovariectomy-affected bone strength. A mechanical densitometric and tomographic (pQCT) study. Bone 17: S373-S378
8. Currey JD (1984). What should bones be designed to do?. Cale tiss Int 36): S7-S10,(y literatura citada)
9. Di Masso R, Font MT, Capozza RF, Detarsio G, Sosa F, Ferretti (1997). Long-bone biomechanics in mice selected for body conformation. bone 20: 539-545
10. Favus Mj (1996). Primer on the metabolic bone diseases and disorders of mkineral metabolism. Raves press, New York
11. Ferretti JL, Audisio E, Tessaro RD, Galassi C, Masoni A, Puche RC (1985). Effect of cholecalciferol, 25 (HO) cholecalciferol and 1,25-(HO)2- cholecalciferol on yhe mechanical proporties of femurs of rachitic chicks fed a P-deficient diet. Nutr. Rep. Ent 37:608-612
12. Ferretti JL, Tessaro RD, Audisio EO, Galassi CD (1985). Long-term effects of high or low CA intakes and of lack of parathyroid function on rat mechanics. Calc Tiss Int 37: 608-612
13. Ferretti JL, Delgado CJ, Ghersevich S, Augsburger S (1987). Biomechanical description of corticoid osteoporosis in rat long bones. En: Calcium Regulation and bone mechabolism: Basisc and Clinical Aspects, Vol. 9 Cohn DV, Martin TJ, Meunier PJ (eds), p668. Elsevier, Amsterdam
14. Ferretti JL, Vazquez SO, Delgado CJ, Capozza R, Cointry G (1992). Biophasic dose-response curves of cortisol effects on rat diaphyseal bone biomechanics. Calc Tiss Int 50: 49-54
15. Ferretti JL, Spiaggi EP, Capozza R, Cointry G, Zanchetta jR (1992). Interrelationships between geometric and mechanical properties of long bones from three rodent species with very different biomass. Phylogenetic implications. J Bone Miner Res 7:S423-S425
16. Ferretti JL, Capozza R, Mondelo N, Montuori E, Zanchetta JR (1993). Determination of femur structural properties by geometric and material variables as a function of body weight in rats. Evidence of a sexual dimorphism. Bone 14:265-270
17. Ferretti JL, Gaffuri OH, Capozza R, Mondelo N, Zanchetta JR (1993). Interrelationships between densitometrical, geometric and mechanical properties of rat femurs. Inferences concerning mechanical regulation of bone modeling. J Bone Miner Res 8:1389-1396
18. Ferretti JL, Capozza R, Tessaro RD, Delgado, zanchetta JR (1995). Monophasic dose-response curves of metamethasone on geometric and mechanical proporties of rat femurs diaphyses in growning rats. Bone: 16:103-108
19. Ferretti JL, Gaffuri OH, Capozza RF, Cointry GR, Bozzini C, Olivera MI, Zanchetta JR, Bozzini C (1995). Dexamethasone effects on structural, geometric and material properties of rat femur diaphyses as described by peripheral quantitative computerized tomography (pQCT) and bending tests. Bone 16, 119-124
20. Ferretti JL, Mondelo N, Capozza RF, Cointry GR, Zanchetta JR, Montuori E (1995). Effects of large doses of opadronate (dimethyl-pamidronate) on mineral density, cross-sectional architecture, and medical properties of rat femurs. Bone, 16: 285S-293S
21. Ferretti JL (1995). Perspectives of pQCT technology associated to biomechanical studies in skeletal research empoying rat models. Bone 17: 353S-364S
22. Ferretti JL, Frost HM, Gasser J, High WB, Jee WSS, Jerome C, Mosekilde L, Thompson DD (1995). Perspectives on osteoporosis research: Its focus and some insights from a new paradigm. Calc Tiss Int; 57: 399-404
23. Ferretti JL (1995). Effects of bisphosphonates on bone biomechanics. En: Bisphosphonate on Bones. Bijvoet OLM, Canfield R, Fleisch H, Russell RGG (eds), pp 211-229. Elsevier, Amsterdam
24. Ferretti JL, Capozza RF, Zanchetta JR (1996). Mechanical validation of atomographic (pQCT) index for the noninvasive assessment of rat femur bending strength. Bone 18: 97-102
25. Ferretti JL (1997). Biomechanical properties of bone. En: Bone Densitometry and Osteoporosis. Genant HK, Gugliemi G, Jergas M (eds), pp. 143-161. Springer Verlag. Berlin
26. Ferretti JL (1997). Noninvasive assessment of bone architecture and biomechanical properties in animals and humans employing pQCT technology. J Jap Soc Bone Morphom, 7: 115-125
27. Ferretti JL, Schiessl H, Frost HM (1998). On new opportunities for absorptiometry. J Clin Densitom, 1: 41-53
28. Ferretti JL, Capozza RF, Cointry GR, Garcia SL, Plotkin H, Alvarez Filgueira ML, Zanchetta JR (1998). Gender-related differences in the relationships between densitometric values of whole-body bone mineral content and lean mass in humans between 2 and 87 years of age. Bone, 22, 683-690
29. Ferretti, JL (1999). Peripheal quantitative computed tomography (pQCT) for evaluating structural and mechanical properties of small bone. En: Practical Guide for Mechanical Testing of Bone. An YH, Draughn RA (eds), CRC Press, Boca Raton, USA
30. Frost HM (1987). The mechanostat: A proposed pathogenic mechanism of osteoporoses and the bone mass effects of mechanical and nonmechanical agents. Bone Miner, 2: 73-85
31. Frost HM (1996). Perspectives: A proposed general model of mechanostat (suggestions from a new paradigm). Anat Rec, 244: 139-147
32. Frost HM (1997). Defining osteopenias and osteoporoses: Another view (with insights from a new paradigm). Bone, 20: 385
33. Frost HM, Ferretti JL, Jee WSS (1998). Some roles of mechanical usage, muscle strength, and the mechanostat in skeletal physiology, disease, and research. Calc Tiss Int, 62: 1-7
34. Kanis J, Melton J, Christiansen C, Johnston CC, Khaltaev N (1994). The diagnosis of osteoporosis. J Bone Miner Res, 9: 1137-1141
35. Ma YF, Ferretti JL, Capozza RF, Cointry GR, Alippi R, Zanchetta RJ, Jee WSS (1995). Effects of on/off anabolic hPTH and remodeling inhibitors on methaphyseal bone of immobilized rat femurs. Tomographical (pQTC) description and correlation with histomorphometric changes in tibial cancellous bone. Bone 17: S321-S328
36. Marotti G, Palazzini S, Palumbo C, Ferretti M (1996). Ultrastructural evidence of the existence of a dendritic network throughout the cells of the osteogenic lineage: the novel concept of wiring, and volume transmission in bone (Abstract). Bone 19 (S3): 151
37. Martin RB (1991). Determinants of the mechanical properties of bone. J Biomech, 24 (S1): 79-88
38. Ott SM, Parfitt AM, Raisz LG, Biewener J (1993). When bone mass fails to predict bone failure. Calc Tiss Int, 53 (S1): S7-S13
39. Rubin CT, McLeod KJ (1996). Inhibition of ostepenia by biophysical intervention. En: Osteoporosis. Marcus R (ed.), pp. 351-371. Academic, New York
40. Schiessl H, Ferretti JL, Tysarczyk-Niemeyer G, Willnecker J (1996). Noninvasive Bone Strength Index as analyzed by periphereal quantitative computed tomography (pQCT). En: Paediatric Osteology. New Developments in Diagnosis and Therapy. Schnau E (ed), pp. 141-145, Elsevier, Amsterdam
Cita Original
Ferretti José Luis. Hacia una Nueva Fisiopatología de las Osteopatías Fragilizantes. Resúmenes del Simposio Internacional de Actualización en Ciencias Aplicadas al Deporte, 131-138, 1999
Cita en PubliCE
José L Ferretti (1999). Hacia una Nueva Fisiopatología de las Osteopatías Fragilizantes. .https://g-se.com/hacia-una-nueva-fisiopatologia-de-las-osteopatias-fragilizantes-807-sa-457cfb2718abb1