Mecanismos Moleculares de Absorción de Glucosa Muscular en Respuesta al Ejercicio de la Fuerza: Una Revisión
Jymmys Lopes dos Santos1,2, Silvan Silva de Araujo2, Charles dos Santos Estevam1, Clésio Andrade Lima3, Carla Roberta de Oliveira Carvalho4, Fábio Bessa Lima4, Anderson Carlos Marçal51 Departamento de Fisiología, Universidad Federal de Sergipe, Brasil
2 Facultad Mauricio de Nassau – Aracaju/SE, Brasil
3 Facultad Ages – Paripriranga/BA, Brasil
4 Departamento de Fisiología y Biofísica, Universidad de São Paulo, Brasil
4 Departamento de Morfología, Universidad Federal de Sergipe, Brasil
Resumen
La evidencia muestra que el ejercicio físico llevado a cabo sistemáticamente promueve numerosos beneficios para la salud, incluyendo mejoras en la homeostasis de la glucosa. De todos los tipos de actividad física, el entrenamiento de la fuerza es efectivo en la promoción de una mayor fuerza, velocidad, potencia, hipertrofia y efectos metabólicos, como el aumento de la sensibilidad a la insulina y el control en el metabolismo del glucógeno. Dado que aún existen lagunas que requieren atención con respecto a los mecanismos moleculares que conducen a la señalización efectiva de la insulina, este documento intenta revisar los efectos y las implicaciones del ejercicio de la fuerza en las principales actividades de la proteína intracelular. En este sentido, nos propusimos comprender el papel del entrenamiento de la fuerza en la actividad de IRS1, IRS2, PI3K, Akt/PKB y GLUT4, lo que puede conducir al desarrollo de posibles intervenciones futuras para mejorar el rendimiento físico y mitigar los efectos de condiciones metabólicas alteradas tales como obesidad, resistencia a la insulina y diabetes mellitus tipo 2.
Palabras Clave: Resistencia a la Insulina, Señalización de Insulina, Proteínas Intracelulares, Ejercicio de la Fuerza
INTRODUCCIÓN
El ejercicio físico es una actividad que se realiza a través de repeticiones sistemáticas de movimientos orientados que se asocian con un mayor consumo de oxígeno debido a la demanda muscular, por lo tanto, generan trabajo. El entrenamiento aeróbico se ha asociado con varios beneficios relacionados con la salud y la mejora en el rendimiento cardiovascular, induciendo respuestas fisiológicas inmunológicas y endócrinas agudas significativas (36,40). Por otro lado, varios autores (5,17,20,48) han informado la importancia de la práctica del ejercicio de la fuerza debido a sus importantes efectos sobre la hipertrofia muscular, el aumento de la fuerza y la calidad muscular en individuos sanos y diabéticos.
En particular, el entrenamiento progresivo de la fuerza se asocia con cambios en individuos con diabetes avanzada con masa corporal magra reducida, actividad disminuida de la enzima glucógeno sintasa (GS) y conversión de fibras musculares tipo 2 a tipo 1 antes de la resistencia a la insulina (17). Si bien existe consenso en cuanto a que tanto los ejercicios aeróbicos como los de fuerza aumentan la sensibilidad a la insulina (6,33), el mecanismo exacto por el cual esto sucede en asociación con cada una de estas modalidades de ejercicio no ha sido del todo definido.
En este contexto, Richter y Ruderman (42) y Sarvas et al. (44) han informado sobre la importancia del estado de energía celular durante el ejercicio de la fuerza para inducir la activación de AMP quinasa (AMPK). Una enzima filogenéticamente conservada, la AMPK se produce en organismos unicelulares y multicelulares (principalmente mamíferos). Es muy sensible al estrés energético celular que se regula alostéricamente al aumentar la concentración de AMP y la fosforilación previa por AMPK. Ha y sus colegas (20) indican que la activación de AMPK desencadena la translocación de vesículas de transportadores de glucosa (GLUT4) y los procesos catabólicos con la liberación de energía para la síntesis y, concomitantemente, la inhibición de los procesos anabólicos que consumen ATP. Como resultado, la célula aumenta la absorción de glucosa independiente de la insulina y la oxidación de la glucosa de los ácidos grasos en la matriz mitocondrial, respectivamente (Figura 1).
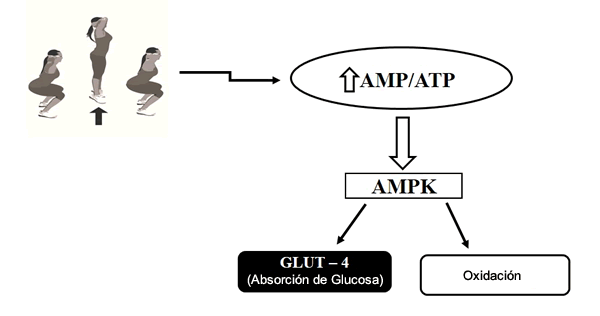
Figura 1. Proceso de Activación de AMPK Inducido por el Ejercicio de la Fuerza y sus Consecuencias Metabólicas, el Aumento de Absorción Celular de GLUT4 y la Oxidación de Ácidos Grasos. Los detalles de este proceso se describen en el texto.
Por el contrario, la inactividad física es un factor de riesgo para el desarrollo de la diabetes tipo 2 y la obesidad (2,30). La estrecha asociación de los cambios intracelulares observados con la inactividad física y la obesidad parece ser una función de la resistencia a la insulina (14). Por lo tanto, dado lo anterior, esta revisión examina las vías moleculares de absorción de glucosa muscular en respuesta al ejercicio de la fuerza. También nos propusimos comprender los mecanismos moleculares que implican la actividad del receptor de insulina (IR), los sustratos IR 1 y 2 (IRS1/2), la proteína fosfoinositol-3-quinasa (PI3K) y la proteína quinasa B (Akt/PKB), y la regulación del transportador de glucosa isoforma 4 (GLUT4) en respuesta al ejercicio de la fuerza agudo y crónico.
MÉTODOS
Los datos para este estudio provienen de las siguientes bases de datos: (a) SciElo, “the Scientific Electronic Library Online”; (b) PubMed/MEDLINE, la Biblioteca Nacional de Medicina; (c) Scopus; y (d) ScienceDirect. Se utilizó una combinación de términos en portugués e inglés, que incluyen ejercicio de la fuerza, resistencia a la insulina, y señalización de insulina, así como los descriptores, PI3K, Akt/PKB y GLUT4. La búsqueda incluyó estudios publicados hasta 2017. Utilizando las palabras clave anteriores, se recuperaron 100 artículos, de los cuales 50 correspondían al propósito de esta revisión, que comprendía los ajustes promovidos por el ejercicio de la fuerza en la vía de señalización de insulina intracelular y las proteínas descendentes en esta vía (IR , IRS, PI3K, Akt/PKB, GLUT4).
RESULTADOS
Activación del Receptor de Insulina y Sustratos del Receptor de Insulina
La hormona insulina se une a su receptor (IR) ubicado en la membrana celular, que desencadena una cascada de señalización que se completa en los efectores descendentes de la vía (15). El IR es una glicoproteína ubicuamente expresada en todos los tejidos celulares que consta de dos subunidades α y dos subunidades β. La unión de la insulina a la subunidad alfa de la IR (Figura 1) promueve la transfosforilación de residuos de tirosina (Tyr) en la subunidad beta del receptor (que se encuentra en la región intracelular), que media la fosforilación de IRSs (13,32). Estos sustratos desempeñan un papel esencial en la transmisión de la señalización intracelular a través de su interacción con la proteína adaptadora Grb2 mediante un reconocimiento específico de los dominios SH2 y SH3, que caracteriza un efecto pleiotrópico de la insulina (49).
En el músculo esquelético, el IRS-1 ocupa un papel esencial en la absorción de la glucosa. Cuando los residuos de tirosina en el IRS-1 se fosforilan, el IRS-1 se convierte en el sustrato principal después de la activación del IR en el músculo esquelético, promoviendo la translocación de vesículas que contienen GLUT4 a la membrana (9,35,41). Varios residuos de serina en la proteína IRS-1 son un objetivo de fosforilación después de la activación de IR, incluidos Ser612, Ser632 y Thr446, lo que da como resultado la transducción de señal intracelular (11). Sin embargo, en condiciones de desequilibrio metabólico como la obesidad y la resistencia a la insulina, la activación del inhibidor del factor nuclear kappa-B quinasa (IKKβ) induce la fosforilación en el residuo Ser307, lo que reduce la absorción de glucosa (21).
En general, la fosforilación de residuos de tirosina en la isoforma IRS-1 es seguida por la activación de la proteína PI3K y, posteriormente, Akt/PKB - moléculas importantes en la absorción de glucosa (11). Por lo tanto, los niveles de fosforilación de IRS-1 determinan la efectividad de la insulina en el proceso de absorción de glucosa en el músculo esquelético. Araki y colegas (4) indicaron que los ratones con supresión del gen IRS-1 tienen retraso en el crecimiento intrauterino y postnatal asociado a una resistencia moderada a la insulina. Además de estas consecuencias, Najeeb y Zou (37) destacaron los déficits de desarrollo y los efectos sobre la homeostasis de la glucosa.
A pesar de los estudios citados anteriormente, todavía existe cierta controversia con respecto a los cambios en la expresión de IRS inducida por el ejercicio. Se informó que la expresión de IRS-1 aumentó un día después del entrenamiento aeróbico exhaustivo y disminuyó 5 días después (31); mientras que, Aguirre et al. (1) no encontraron cambios en la expresión de IRS-1 e IRS-2 siete días después del ejercicio. De acuerdo con estos estudios, Aoi et al. (3) también demostraron que los ratones sometidos al ejercicio excéntrico y aeróbico exhaustivo tenían alteración de la fosforilación de residuos de tirosina IRS-1 y de la señalización de PI3K/Akt en el músculo esquelético en comparación con un grupo control no sometido al ejercicio, lo que resulta en una menor translocación de GLUT4 a la membrana. Esta evidencia sugiere que los efectos promovidos por el ejercicio aeróbico en la expresión de IRS-1 dependen de diferentes condiciones, como la duración y la intensidad del ejercicio. Un estudio de Jorge et al. (28) ha demostrado un aumento de la absorción de glucosa en humanos sometidos a diferentes modalidades de entrenamiento. Los autores sometieron a pacientes con sobrepeso y obesos a diferentes tipos de entrenamiento (de fuerza, aeróbico y combinado) y verificaron una mayor expresión de IRS-1 en los grupos implicados en el entrenamiento de la fuerza y el combinado. Por lo tanto, la expresión aumentada de IRS-1 y la consiguiente fosforilación de sus residuos de tirosina explican una mayor absorción de glucosa que persistió durante varias horas después del ejercicio.
Actividad de PI3K
Las enzimas PI3K son miembros de una familia de quinasas heterodiméricas intracelulares, compuestas de subunidades reguladoras y catalíticas que reclutan lípidos como segundos mensajeros. Cuando se activan, estas moléculas modulan varias funciones intracelulares importantes, como la inhibición de la apoptosis, el crecimiento, la diferenciación, el metabolismo, la quimiotaxis y el tránsito de vesículas celulares (10,39).
Las diversas funciones de PI3K están asociadas con sus isoformas, que constan de tres clases distintas categorizadas de acuerdo con su estructura y mecanismos de activación. Las isoformas de clase I son las isoformas más estudiadas, debido a su participación en la señalización del receptor de tirosina quinasa. A través de proteínas adaptadoras, como IRS, convierte la membrana fosforilada de isoformas clase I, fosfatidilinositol 4,5-bisfosfato (PIP2) en fosfatidilinositol 3,4,5-trifosfato (PIP3) (28). La subfamilia de clase I consta de cuatro subunidades catalíticas: tres subunidades IA (p110α, p110β y p110δ) y una subunidad de clase IB (p110γ). Las subunidades reguladoras de clase I que son esenciales para la interacción con receptores tirosina quinasas son p85α, p85β, p55α, p55γ y p50α. La clase II comprende tres isoformas (PI3K-C2α, PI3K-C2β y PI3K-C2γ) activadas por los receptores unidos a la proteína G. Finalmente, la isoforma de clase III también se llama Vps34 (3,7).
La subunidad p85 de PI3K juega un papel importante en las interacciones de mediación por la activación de la insulina y los receptores del factor de crecimiento similar a la insulina (IGF) mediante IRS-1 e IRS-2 (30,31) y la activación de la proteína piruvato deshidrogenasa quinasa (PDK), dando como resultado la fosforilación de Akt/PKB. Esta cascada es esencial para la fosforilación de la proteína quinasa diana de mamíferos (mTOR), que mejora la síntesis de proteínas activando p70S6K y 4E-BP1 y, en consecuencia, regula la hipertrofia del músculo esquelético (9,32,54). Por otro lado, Hamilton et al. (22) no encontraron cambios asociados con el ejercicio de la fuerza en la cascada de PI3K/Akt con la activación posterior de mTOR, a pesar del bloqueo del homólogo de fosfatasa y tensina (PTEN), un regulador negativo de la interacción entre insulina y PI3K. Los informes (26,29,40) han señalado una asociación directa entre el ejercicio regular sobre la disponibilidad de glucosa en el músculo esquelético, así como una mayor sensibilidad a la insulina con la concomitante actividad PI3K mediada por la insulina.
Como lo demostraron Melo et al. (36), la activación de la cascada descendente de PI3K potencia la hipertrofia concéntrica de miocitos cardíacos en ratas sometidas al entrenamiento de la fuerza. Esto es consistente con el estudio de Luo et al. (34), en el que una supresión de PI3K dañó notablemente la absorción de glucosa y contribuyó al desarrollo de atrofia muscular. En pacientes con diabetes tipo 2 que se someten a un programa de ejercicio de la fuerza, hay aumentos en la actividad de PI3K, en el contenido de Akt, y la actividad GLUT4 y GS.
Activación de Akt/PKB durante el Ejercicio
Akt/PKB es una serina/treonina (Ser/Thr) quinasa activada por PI3K. También denominadas RAC-PK ("relacionadas con las quinasas A y C"), las isoformas Akt comprenden una familia de tres miembros - Akt1 (PKB-α), Akt2 (PKB-β) y Akt3 (PKBγ) - y sus actividades son muy susceptibles a alteraciones metabólicas como la obesidad, la resistencia a la insulina y la diabetes tipo 2 (18). Las isoformas Akt1 y Akt2 se expresan predominantemente en el músculo esquelético, el cerebro, el corazón y los pulmones; mientras que, Akt3 se expresa principalmente en el cerebro. Akt/PKB fosforila proteínas diana reguladoras que son importantes para la absorción de glucosa e inhibe la apoptosis (27,35,36).
Además de los efectos anteriores, la Akt/PKB puede activarse por mecanismos independientes de la señalización de PI3K, el tratamiento con la hormona de crecimiento y el aumento de la concentración intracelular de iones de calcio; estos efectos se deben en parte al aumento en la concentración observada de cAMP durante la contracción muscular (12,46). Además, hay informes de una mayor actividad y fosforilación de Akt/PKB Ser/Thr relacionada con la hipertrofia muscular a través de la activación de mTOR y la inhibición de la glucógeno sintasa quinasa 3β (GSK-3β), lo que lleva a cambios importantes en el grado de sensibilidad a la insulina y concentraciones circulantes de esta hormona (43). Un estudio publicado por Hers et al. (24) ha demostrado que la regulación de Akt/GSK-3β y Akt/mTOR en el músculo esquelético humano es fundamental en la hipertrofia muscular; una vía que es paralela a la disminución de FOXO1, un factor de transcripción asociado con la estimulación de la atrofia muscular (50).
Al corroborar las actividades de Akt mencionadas anteriormente, Choi y Bum Kim (12) encontraron que los ratones knockout Akt1 presentan un patrón de crecimiento corporal alterado. Mientras tanto, Ogasaswara y sus colegas (38) enfatizan el papel efector de Akt en la hipertrofia muscular a través de una vía que involucra mTOR sensible a la rapamicina. Aunque, como ya se confirmó en un estudio anterior (43), esta vía de señalización es un estímulo contráctil dependiente. Curiosamente, discrepando con esta línea de evidencia, Zanchi y Lancha (53) enfatizaron que tanto el ejercicio aeróbico como el de la fuerza pueden no causar la fosforilación de Akt con la activación posterior. Esto puede ocurrir por varias razones aún no aclaradas, principalmente por la existencia de múltiples isoformas de Akt en el músculo esquelético, especialmente cuando no predomina Akt1.
Vía Akt/PKB-GLUT4
Los transportadores de glucosa de diferentes isoformas permiten el transporte de glucosa al entorno intracelular. En las células musculares, las vesículas que contienen GLUT4 se trasladan a la membrana plasmática después de la estimulación con insulina para promover la absorción de glucosa en la célula. La interacción de la insulina con su receptor en la membrana es el principal estímulo extracelular que determina la translocación de GLUT4 y promueve la absorción de glucosa en las células del músculo esquelético, aunque estudios recientes han demostrado la disponibilidad de moléculas de señalización en GLUT4 a través de una vía independiente de insulina y otros enlaces. (Figura 2) (45,52).
El aumento de la sensibilidad a la insulina culmina en efectos que se deben en parte al aumento en el grado de fosforilación de Akt/PKB (23,47) por PI3K, que permite la fosforilación de su sustrato AS160 (proteína activadora de GTPasa Rab [GAP]) Osorio -Fuentealba et al. (40). En las fibras musculares, la estimulación contráctil promueve cambios moleculares que desencadenan la activación de dos vías independientes, que incluyen mTOR y GSK-3β, que conducen a la hipertrofia del músculo esquelético (43).
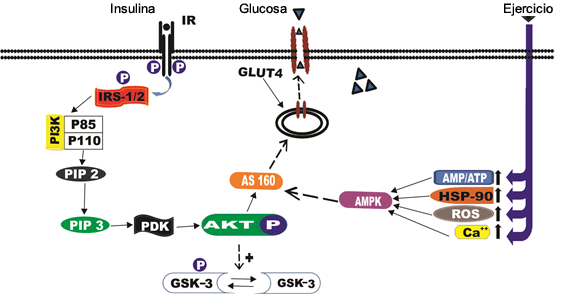
Figura 2. Insulina Intracelular y Vías de Señalización Independientes de Insulina Desencadenadas por el Ejercicio. La Unión de la Insulina a la Subunidad α del Receptor de Insulina Provoca que el Receptor se Autofosforile, lo que Conduce a la Fosforilación de Sustratos Intracelulares, como el IRS. La Fosforilación es Seguida por la Activación de Vías de Señalización Intracelular tales como la Vía PI3K, Akt/PKB y la cascada AMPK.
Esta evidencia sugiere que el ejercicio puede activar las vías moleculares que movilizan las vesículas de GLUT4 independientes de la insulina. Sin embargo, los estudios han demostrado que los beneficios del ejercicio de la fuerza en la expresión y translocación de GLUT4 resultan del reclutamiento de moléculas relacionadas con el estrés celular promovido por el proceso contráctil, como los de la vía dependiente de señalización de AMPK (Figura 2). Esta enzima puede ser activada por Ca++/ proteína quinasa dependiente de calmodulina (CaMK), quinasa hepática B1, proteína quinasa C (PKC), proteína de choque térmico 90 (HSP90) y óxido nítrico (NO) (3,5,8,16,25).
En otra línea de observación (5), el aumento de la actividad de AMPK en respuesta a una mejor tolerancia a la glucosa también se ha asociado con el entrenamiento físico. Andersen et al. (2) han observado en el músculo esquelético una transcripción aumentada del ARNm de GLUT4 y varias otras proteínas implicadas en la transducción de señales de insulina y el metabolismo de la glucosa, tales como PI3K, Akt, AMPK y mTOR después de la práctica del ejercicio de la fuerza.
Una vez activado, AMPK influye en varios objetivos moleculares que desencadenan diferentes procesos celulares largamente dilucidados, incluidos el metabolismo de los lípidos y los carbohidratos, la señalización celular, la fosforilación de IRS y el transporte iónico (17, 51). La secuencia de señalización de AMPK implica una fosforilación directa del sustrato AS160, que controla el reciclado de las vesículas GLUT4 y su translocación de membrana (37), lo que resulta en la absorción de glucosa independiente de la insulina.
CONCLUSIONES
En la presente revisión, presentamos evidencia importante sobre los efectos del ejercicio de la fuerza en la modulación de las vías de señalización intracelular que conducen a la absorción de glucosa. En este sentido, la interacción del IR y sus sustratos (IR/IRS-1/IRS-2), PI3K y Akt conduce a la síntesis de proteínas y la translocación de GLUT4 a la membrana. Las vías de señalización desencadenadas por la insulina se potencian mediante la práctica regular del ejercicio de la fuerza. La absorción de glucosa también es evidente por la activación de AMPK por marcadores moleculares resultantes del estrés celular en respuesta al ejercicio de la fuerza. Si bien estos mecanismos contribuyen directamente a la dilucidación de las vías que no se han descrito, pueden estar involucrados en respuesta al ejercicio físico de la fuerza. Esta revisión ofrece perspectivas para el desarrollo de posibles intervenciones para la mejora del rendimiento físico y la atenuación de los efectos de trastornos metabólicos como la obesidad, la resistencia a la insulina y la diabetes tipo 2.
AGRADECIMIENTOS
CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) para las becas de maestría.
Dirección de correo: Jymmys Lopes dos Santos, Post-graduate Program in Biotechnology – University Federal de Sergipe ( UFS). Avenida Marechal Rondon, S/N - Rosa Elze, São Cristóvão - SE, 49100-000, São Cristovão Sergipe, Brazil. Email: jymmyslopes@yahoo.com.br
Referencias
1. Aguirre V, Uchida T, Yenush L, Davis R, White MF. (2000). The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J Biol Chem; 24;275:(12)9047-9054.
2. Andersen JL, Schjerling P, Andersen LL, Dela F. (2003). Resistance training and insulin action in humans: Effects of de-training. J Physiol. 2003;(15);551:1049-1058.
3. Aoi W, Naito Y, Tokuda H, Tanimura Y, Oya-ito T, Yoshikawa T. (2012). Exercise-induced muscle damage impairs insulin signaling pathway associated with IRS-1 oxidative modification. Physiol Res. 2012;61(1):81-88.
4. Araki E, Lipes MA, Patti ME, Bruning JC, Haag B 3rd, Johnson RS, et al. (1994). Alternative pathway of insulin signaling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372:186-190.
5. Borges GA, Araújo SFM, Cunha RM. (2010). Os benefícios do treinamento resistido para portadores de diabetes mellitus tipo II. Lecturas Educación Física y Deportes. 2010; (12):151:1-1.
6. Brooks N, Layne JE, Gordon PL, Roubenoff R, Nelson ME, Castaneda-Sceppa C. (2007). Strength training improves muscle quality and insulin sensitivity in Hispanic older adults with type 2 diabetes. Int J Med Chem Sci. 2007;4(1):19-27.
7. Cain RJ, Ridley AJ. (2009). Phosphoinositide 3-kinases in cell migration. Biol Cell. 2009;101 (1):13-29.
8. Calle MC, Fernandez ML. (2010). Effects of resistance training on the inflammatory response. Nutr Res Pract. 2010;(4)259-269.
9. Carvalheira JBC, Zecchin HG, Saad MJA. (2002). Vias de Sinalização da Insulina. Arq Bras Endocrinol Metab. 2002;(46):419-425.
10. Chibalin AV, Yu M, Ryder JW, Song XM, Galuska D, Krook A, et al. (2000). Exercise-induced changes in expression and activity of proteins involved in insulin signal transduction in skeletal muscle: Differential effects on insulin receptor substrates 1 and 2. Proc Natl Acad Sci USA. 2000;(97):38-43.
11. Chiu TT, Jensen TE, Sylow L, Richter EA, Klip A. (2011). Rac1 signalling towards GLUT4/glucose uptake in skeletal muscle. Cell Signal. 2011;23(10):1546-1554.
12. Choi K, Bum Kim YB. (2010). Molecular mechanism of insulin resistance in obesity and type 2 diabetes. Korean J Intern Med. 2010;(25):119-129.
13. Christ-Roberts CY, Pratipanawatr T, Pratipanawatr W, Berria R, Belfort R, Kashyap S, et al. (2004). Exercise training increases glycogen synthase activity and GLUT4 expression but not insulin signaling in overweight nondiabetic and type 2 diabetic subjects. Metab. 2004;53(9):1233-1242.
14. Ciolac EG, Guimarães GV. (2004). Exercício físico e síndrome metabólica. Rev Bras Med Esporte. 2004;(10)4:319-324.
15. Ciolac EG, Guimarães GV. (2002). Importância do exercício resistido para o idoso. Ver Soc Cardiol Est São Paulo. 2002;(12):S15-26.
16. Dale S, Wilson WA, Edelman AM, Hardie DG. (1995). Similar substrate recognition motifs for mammalian AMP-activated protein kinase, higher plant HMG-CoA reductase kinase-A, yeast SNF1, and mammalian calmodulin-dependent protein kinase I. FEBS Lett. 1995;361(2-3):191-195.
17. Dela F, Mikines KJ, Larsen JJ, Galbo H. (1999). Glucose clearance in aged trained skeletal muscle during maximal insulin with superimposed exercise. J Appl Physiol. 1999; (10):262-267.
18. Downward J. (1998). Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;(10):2:262-267.
19. Forjaz CLM, Rezk, CC, Melo CM, Santos DA, Teixeira L, et al. (2003). Exercício resistido para o paciente hipertenso: Indicação ou contra-indicação. Revista Brasileira de Hipertensão, Ribeirão Preto. 2003;(10):2:119-124.
20. Ha J, Guan KL, Kim J. (2015). AMPK and autophagy in glucose/glycogen metabolism. Mol Aspects Med. 2015;(46):46-62.
21. Haber PE, Curi R, Carvalho CRO, Carpinelli AR. (2001). Secreção da Insulina: Efeito Autócrino da Insulina e Modulação por Ácidos Graxos. Arq Bras Endocrinol Metab. 2001;(45):219-227.
22. Hamilton DL, Philp A, MacKenzie MG, Baar K. (2010). A Limited Role for PI(3,4,5)P3 regulation in controlling skeletal muscle mass in response to resistance exercise. PLoS One. 2010;(5):7;11624.
23. Hawkins PT, Anderson KE, Davidson K, Stephens LR. (2006). Signaling through class I PI3Ks in mammalian cells. Biochem Soc Trans. 2006;(34):647-662.
24. Hers I, Vincent EE, Tavaré JM. (2011). Akt signalling in health and disease. Cell Signal. 2011; 23(10):1515-1527.
25. Holten MK, Zacho M, Gaster M, Juel C, Wojtaszewski FFP, Dela F. (2004). Strength training increases insulin-mediated glucose uptake, GLUT4 content, and insulin signaling in skeletal muscle in patients with Type 2 diabetes. Diabetes. 2004;(53):294-305.
26. Houmard JA, Shaw CD, Hickey MS, Tanner CJ. (1999). Effect of short-term exercise training on insulin-stimulated PI 3-kinase activity in human skeletal muscle. Am J Physiol. 1999;277:(6):E1055-1060.
27. Jones PF, Jakubowic ZT, Pitossi FJ, et al. (1991). Molecular cloning and identification of a serine/threonine protein kinase of the second-messenger subfamily. Proc Natl Acad Sci. 1991;15;88(10):4171-4175.
28. Jorge MLMP, Oliveira VN, Resende NM, Paraiso LF, Calixto A, Diniz ALD, et al. (2011). The effects of aerobic, resistance, and combined exercise on metabolic control, inflammatory markers, adipcytokines, and muscle insulin signaling in patients with type 2 diabetes mellitus. Metab. 2011;60(9):1244-1252.
29. Kirwan JP, Aguila DLF, Hernandez JM, Williamson DL, O’Gorman DJ, Lewis R, et al. (2000). Regular exercise enhances insulin activation of IRS-1-associated PI3-kinase in human skeletal muscle. J Appl Physiol. 2000;(88):2:797-803.
30. Kraemer WJ, Ratamess NA. (2004). Fundamentals of resistance training: Progression and exercise prescription. Med Sci Sports Exerc. 2004;(36):4:674-688.
31. Langlais P, Yi Z, Finlayson J, Luo M, Mapes R, De Filippis E, et al. (2011). Global IRS-1 phosphorylation analysis in insulin resistance. Diabetologia. 2011;(54):2878-2889.
32. Lebrasseur NK, Walsh K, Arany Z. (2011). Metabolic benefits of resistance training and fast glycolytic skeletal muscle. Am J Physiol Endocrinol Metab. 2011;300:E3-E10.
33. Lehnen AM, Angelis KD, Markoski MM, Schaan BDA. (2012). Changes in the GLUT4 expression by acute exercise, exercise training and detraining in experimental models. J Diabetes Metab. 2012;S10:002.
34. Luo J, Sobkiw CL, Hirshman MF, Logsdon NM, Li TQ, Goodyear LJ, et al. (2006). Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab. 2006;3;(5):355-366.
35. Machado UB. (1998). Transportadores de Glicose. Arq Bras Endocrinol Metab. 1998;42(6): 413-421.
36. Melo SFS, Amadeu MA, Magalhães CF, Fernandes T, Carmo EC, Barretti DLM, et al. (2011). Exercício de força ativa a via AKT/mTor pelo receptor de angiotensina II tipo I no músculo cardíaco de ratos. Rev Bras Educ Fís Esporte São Paulo. 2011(25):3:377-385.
37. Najeeb AS, Zou MH. (2014). AMPK: A cellular metabolic and redox sensor. A mini-review. Front Biosci (Landmark Ed). 2014;(1):19:447-474.
38. Ogasawara R, Fujita S, Hornberger TA, Kitaoka Y, Makanae Y, Nakazato K, Naokata I. (2016). The role of mTOR signalling in the regulation of skeletal muscle mass in a rodent model of resistance exercise. Scientific Reports. 2016;(6):31142.
39. O'Gorman DJ, Karlsson HK, McQuaid S, Yousif O, Rahman Y, Gasparro D, et al. (2006). Exercise training increases insulin-stimulated glucose disposal and GLUT4 (SLC2A4) protein content in patients with type 2 diabetes. Diabetologia. 2006;49(12):2983-2992.
40. Osorio-Fuentealba C, Ariel E. Contreras-Ferrat, FA, Espinosa A, Li Q, Niu W, Lavandero S, Klip A, Jaimovich E. (2013). Electrical stimuli release ATP to increase GLUT4 translocation and glucose uptake via PI3Kg-Akt-AS160 in skeletal muscle cells. Diabetes. 2013;(62):1519-1526.
41. Pauli JR, Cintra DE, Souza CT, Ropelle ER. (2009). Novos mecanismos pelos quais o exercício físico melhora a resistência à insulina no músculo esquelético. Arq Bras Endocrinol Metab. 2009;53(4):399-408.
42. Richter EA, Ruderman NB. (2009). AMPK and the biochemistry of exercise: Implications for human health and disease. Biochem J. 2009;1;418(2):261-275.
43. Sakamoto K, Arnolds DEW, Ekberg I, Thorell A, Goodyear LJ. (2004). Exercise regulates Akt and glycogen synthase kinase-3 activities in human skeletal muscle. Biochemical and Biophysical Research Communications. 2004:325:419-425.
44. Sarvas JL, Otis JS, Khaper N, Lees SJ. (2015). Voluntary physical activity prevents insulin resistance in a tissue specific manner. Physiol Rep. 2015;3(2):e12277.
45. Seraphim PM, Nunes MT, Giannocco G, Machado UF. (2007). Age related obesity-induced shortening of GLUT4 mRNA poly(A) tail length in rat gastrocnemius skeletal muscle. Mol Cell Endocrinol. 2007;(30):276:80-87.
46. Sharma N, Arias BE, Bhat AD, Sequea DA, Ho S, Croff KK, et al. (2011). Mechanisms for increased insulin-stimulated Akt phosphorylation and glucose uptake in fast- and slow-twitch skeletal muscles of calorie-restricted rats. Am J Physiol Endocrinol Metab. 2011;300:E966-E978.
47. Shaw M, Cohen P, Alessi DR. (1998). The activation of protein kinase B by H2O2 or heat shock is mediated by phosphoinositide 3-kinase and not by mitogen-activated protein kinase-activated protein kinase-2. Biochem J. 1998;(15):336:241-246.
48. Silva PE, Alves T, Fonseca ATS, Oliveira MAN, Machado UF, Seraphim PM. (2011). Physical Exercise improves insulin sensitivity of rats exposed to cigarette smoke. Rev Bras Med Esporte. 2011;17:(3).
49. Taniguchi CM, Emanuelli B, Kahn CR. (2006). Critical nodes in signalling pathways: Insights into insulin action. Nat Rev Mol Cell Biol. 2006;(7):85-96.
50. Tsuchida A, YamauchiT, Ito Y, Hada Y, Maki T, Takekawa S, et al. (2004). Insulin/Foxo1 Pathway regulates expression levels of adiponectin receptors and adiponectin sensitivity. J Biol Chem. 2004;29(16):30817-30822.
51. Volek JS, Feinman RD. (2005). Carbohydrate restriction improves the features of Metabolic Syndrome. Metabolic Syndrome may be defined by the response to carbohydrate restriction. Nutrition & Metabolism. 2005;2:31.
52. Weekes J, Ball KL, Caudwell FB, Hardie DG. (1993). Specificity determinants for the AMP-activated protein kinase and its plant homologue analysed using synthetic peptides. FEBS Lett. 1993;11:22;334(3):335-339.
53. Yaspelkis BB III, Singh MK, Trevino B, Krisan AD, Collins DE. (2002). Resistance training increases glucose uptake and transport in rat skeletal muscle. Acta Physiol Scand. 2002;175:315-323.
54. Zanchi NE, Lancha AH Jr. (2008). Mechanical stimuli of skeletal muscle: Implications on mTOR/p70s6k and protein synthesis. Eur J Appl Physiol. 2008;102(3):253-263.
55. Zhang P, Chen X, Fan M. (2007). Signaling mechanisms involved in disuse muscle atrophy. Med Hypotheses. 2007;(69):310-321.
Cita Original
Santos JL, Araujo SS, Estevam CS, Lima CA, Carvalho CRO, Lima FB, Marcal AC. Mecanismos Moleculares de Absorción de Glucosa Muscular en Respuesta al Ejercicio de la Fuerza: Una Revisión. JEPonline 2017;20(4):200-211.
Cita en PubliCE
Jymmys Lopes dos Santos, Silvan Silva de Araujo, Charles dos Santos Estevam, Clésio Andrade Lima, Carla Roberta de Oliveira Carvalho, Fábio Bessa Lima Anderson Carlos Marçal (2018). Mecanismos Moleculares de Absorción de Glucosa Muscular en Respuesta al Ejercicio de la Fuerza: Una Revisión. .https://g-se.com/mecanismos-moleculares-de-absorcion-de-glucosa-muscular-en-respuesta-al-ejercicio-de-la-fuerza-una-revision-2388-sa-D5ac6df45f3031